DMOA(3,5-二甲基苔色酸)-衍生的杂萜类天然产物,目前已有超过200个化合物的分离报道,具有结构复杂、数量庞大、氧化态丰富、生物活性显著等特点。目前对该类天然产物的合成主要包括simplicissin, berkeleyone A, andrastin D等,Porco,Jr. Maimone,Newhouse, 黎后华,谢志翔等课题组在该领域做出了贡献。
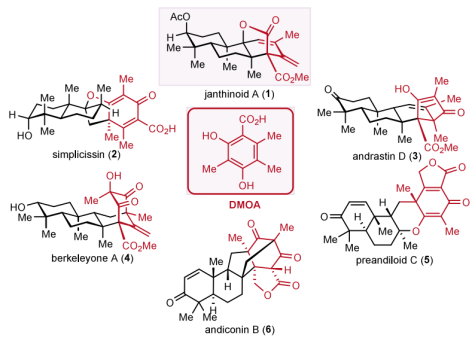
图1. 天然存在的DMOA衍生的混萜类化合物
(图片来源:J. Am. Chem. Soc.)
Janthinoid A为2021年中国农业科学院烟台烟草研究所的张鹏课题组从微紫青霉菌中分离出来的该类型天然产物,该化合物展现出了体内抑制非小细胞肺癌细胞A549的活性,同时在结构上包含了六个手性中心,四个连续季碳手性中心,其中三个季碳中心位于高度官能团化的[3.2.1]桥环内酯结构单元中。
近期,杨震/张仲超团队报道了天然产物Janthinoid A的首次不对称全合成,从商业可得的香叶基丙酮出发,无需保护基,总反应步数为14步,总收率为3.8%。该路线的关键反应包括:1)环氧启动、联烯参与的阳离子多烯环化反应,构建重要杂萜合成中间体烯醛;2)高氯酸铁介导的氧化自由基环化反应,一步构建了刚性的[3.2.1]桥环内酯骨架结构、3个连续季碳手性中心。
逆合成分析如图所示:1中的[3.2.1]桥环内酯结构可以通过氧化自由基环化反应从三羰基底物C得到,C则可以通过HWE反应和缩合反应从已报道的烯醛D构建。在此,作者设计了使用联烯底物E,通过阳离子环化方式构建该化合物。联烯则可通过炔丙醇酯F通过已报道的1,3酰基迁移手段来合成。F则可以通过3步简单的官能团化,从商业可得的香叶基丙酮来制备。

图2.逆合成分析
(图片来源:J. Am. Chem. Soc.)
全合成路线如下:首先,作者从商业可得的香叶基丙酮出发,使用了ent-Corey–Zhang配体,以97%的产率,92%的ee值,完成了底物的不对称双羟化,随后通过分子内SN2反应构建了具有C3位手性为R的环氧化合物,并在乙炔基溴化镁和特戊酰氯的条件下得到炔丙醇酯化合物9。接下来在三氟乙酸铑的条件下通过分子内1,3酰基迁移得到了联烯酯10。对该多烯环化反应进行条件筛选,最优条件为BF3·Et2O/DCM体系,以65%的产率得到了目标烯醛11。一共五步,不对称地得到了该重要杂萜合成中间体。
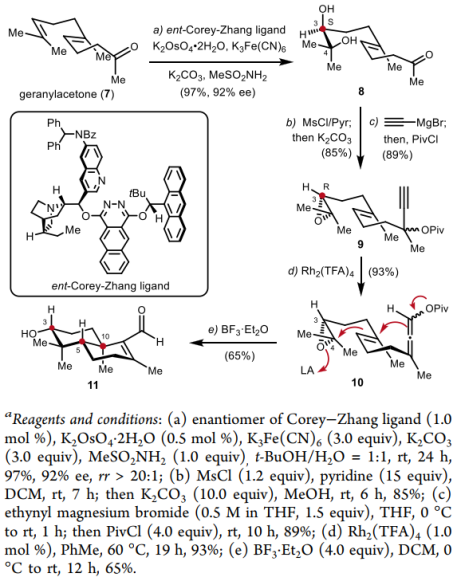
图3. 醛11的合成
(图片来源:J. Am. Chem. Soc.)
在得到该烯醛后,作者对侧链的引入展开了研究。首先将C3位羟基进行氧化为酮,在HWE条件下,区域选择性地得到了纯E式烯酯14,两步收率为69%。随后通过K-selectride对酮羰基进行立体选择性还原,以98%的收率,dr值为4.8:1得到了非对映异构体15,并在氢氧化锂水解的条件下得到了羟基羧酸16。化合物14与16均通过X射线衍射确定其结构。对羟基上乙酯得到羧酸17,2步产率为96%。随后将羧基转化为酰氯,与丙二酸二甲酯缩合,以70%的收率得到三羰基化合物18,进而为后续的氧化自由基环化反应研究提供基础。
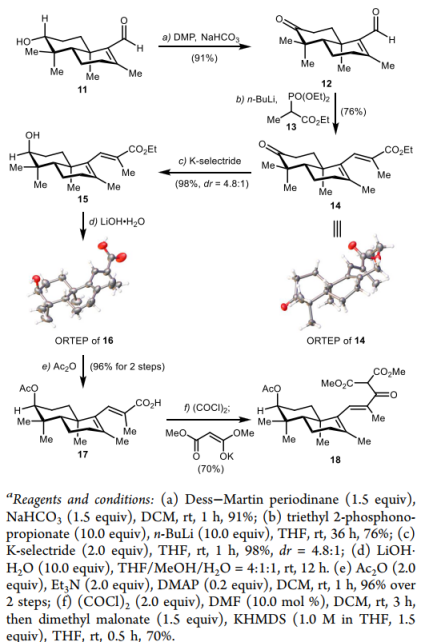
图4. 酮酯18的合成
(图片来源:J. Am. Chem. Soc.)
接下来,作者基于Citterio在1993年氧化自由基环化反应实现烯烃E/Z异构化的报道(Citterio, A. et al. Tetrahedron 1993, 49, 7743−7760.),对关键的氧化自由基环化反应展开了条件探索。首先应用了经典的氧化自由基环化条件Mn(OAc)3/AcOH体系,发现仅能得到底物分解的结果,而将溶剂替换为DCM或MeCN时,则无反应发生。此时作者往体系中加入1当量的三氟乙酸,发生有20%的目标环化产物19生成,并通过X射线衍射确定其结构。随后作者尝试将氧化剂替换为高氯酸铁,发现产率提高至55%,反应温度的升高或降低均会导致产率的降低。替换溶剂为DCM时,由于高氯酸铁在该溶剂中的溶解性较差,反应停止发生。作者也尝试了其他FeIII盐如FeCl3, Fe(acac)3,均未获得较好的反应结果。基于此,该反应成功构建了右侧包含连续3个季碳手性中心在内的拥挤的[3.2.1]桥环内酯骨架结构,完成了Janthinoid A的骨架构筑。
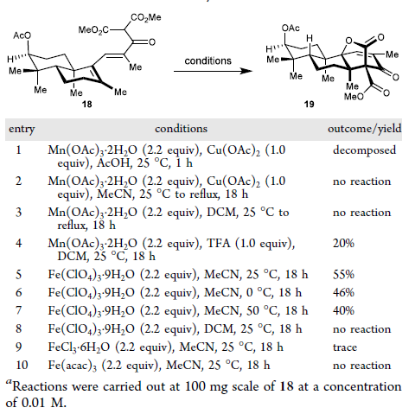
图5. 串联氧化/异构化/环化反应的条件筛选
(图片来源:J. Am. Chem. Soc.)
在合成的最后部分,仅需实现化合物19的C13位的亚甲基化即可完成天然产物的全合成,但是由于该烯酮位于拥挤的立体环境中,且附近具有较为丰富的亲核受体,常见的亚甲基化条件如Wittig, Peterson, Julia, Tebbe, Takai等烯基化反应均未获得成功。最后作者以LaCl3·(LiCl)2为辅剂,甲基锂加成后Burgess脱水,以42%的收率实现了该步亚甲基化反应,完成了天然产物Janthinoid A的首次不对称全合成。
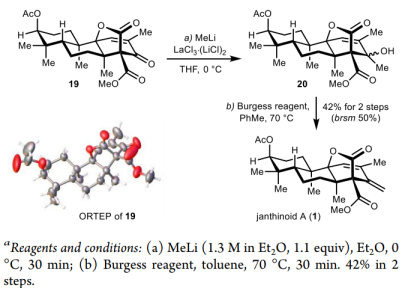
图6. janthinoid A (1)的不对称合成
(图片来源:J. Am. Chem. Soc.)
最后,为了阐明异构化/氧化成环反应机制,作者在PWPB95-D3/def2-QZVPP//ωB97X-D/def-TZVP理论水平下进行了计算化学研究。所有计算均在乙腈溶剂化条件下进行,相关结果展示于下图中。
针对E/Z异构化过程,经筛选得到最优路径为酯羰基氧原子对烯烃的可逆5-exo-trig自由基加成过程。该步骤具有较高的反应速率,过渡态TS1相对于自由基中间体Int1a的活化吉布斯自由能为13.4 kcal/mol。从立体选择性角度分析,凸面加成方式(TS1)比凹面加成(TS1')略微更有利。随后经历快速的C-C单键旋转(TS2),C-O键解离步骤的活化吉布斯自由能为10.2 kcal/mol(过渡态TS3),最终形成中间体Int4a。
Int4a与Int4b之间存在构象平衡。其中酮羰基与酯基共平面构型的Int4a稳定性较低,其自旋密度主要分布在酮羰基氧原子和二烯基团区域。而在Int4b中,O=C(Me)-CO2Me二面角接近90°,此时自旋密度集中于丙二酸酯基团的α-C位点。完成构象转变后,Int4b可顺利通过6-endo-trig生成稳定的中间体Int5。
其他竞争性较弱的路径包括基于3-exo-trig的E/Z异构化过程,其有效活化能计算值为28.8 kcal/mol;以及需要25.3 kcal/mol活化能的4-endo-trig自由基加成过程。这两条路径在反应条件下均表现为能量上不可行。

图7. DFT计算结果
(图片来源:J. Am. Chem. Soc.)
总结
杨震/张仲超团队从商业可得的香叶基丙酮出发,一共通过无保护基的14步转化,完成杂萜天然产物Janthinoid A的首次不对称全合成。关键策略包括:环氧启动、联烯参与的阳离子多烯环化反应构建重要杂萜合成中间体烯醛,和高氯酸铁介导的氧化自由基环化反应完成Janthinoid A右侧 [3.2.1]桥环内酯的构建,为其他具有类似骨架结构的天然产物合成研究提供了新途径。
北京大学的唐福博士研究生和张仲超副研究员为该工作的共同第一作者,宋治霖博士研究生和李元鹤博士对DFT理论计算部分做出了重要贡献。北京大学杨震教授和张仲超副研究员为该文章的共同通讯作者。该工作得到了国家自然科学基金、广东省自然科学基金、深圳市基础研究计划、深港脑科学创新研究院、深圳湾实验室、深圳市杰出人才培养基金的支持。
全文链接:https://pubs.acs.org/doi/10.1021/jacs.4c17480?articleRef=test
Asymmetric Total Synthesis of Janthinoid A
Fu Tang, Zhong-Chao Zhang,* Zhi-Lin Song, Yuan-He Li, Zi-Hao Zhou, Jia-Jun Chen, Zhen Yang.*




